El 29 de febrero se conmemora el Día Mundial de las Enfermedades Poco Frecuentes (EPOF) con el fin de concientizar y promover el desarrollo de políticas que garanticen el acceso a la salud de este tipo de patologías que afectan a 3,6 millones de personas en nuestro país.
Hay descriptas clínicamente 8000 EPOF: crónicas, complejas, progresivas, discapacitantes y, en ciertos casos, potencialmente mortales. El 72 por ciento, es decir 7 de cada 10, son de origen genético, y de ellas, el 70 por ciento se manifiestan al nacer o durante la niñez. En nuestro país una de cada cuatro familias sufre una enfermedad de este tipo.
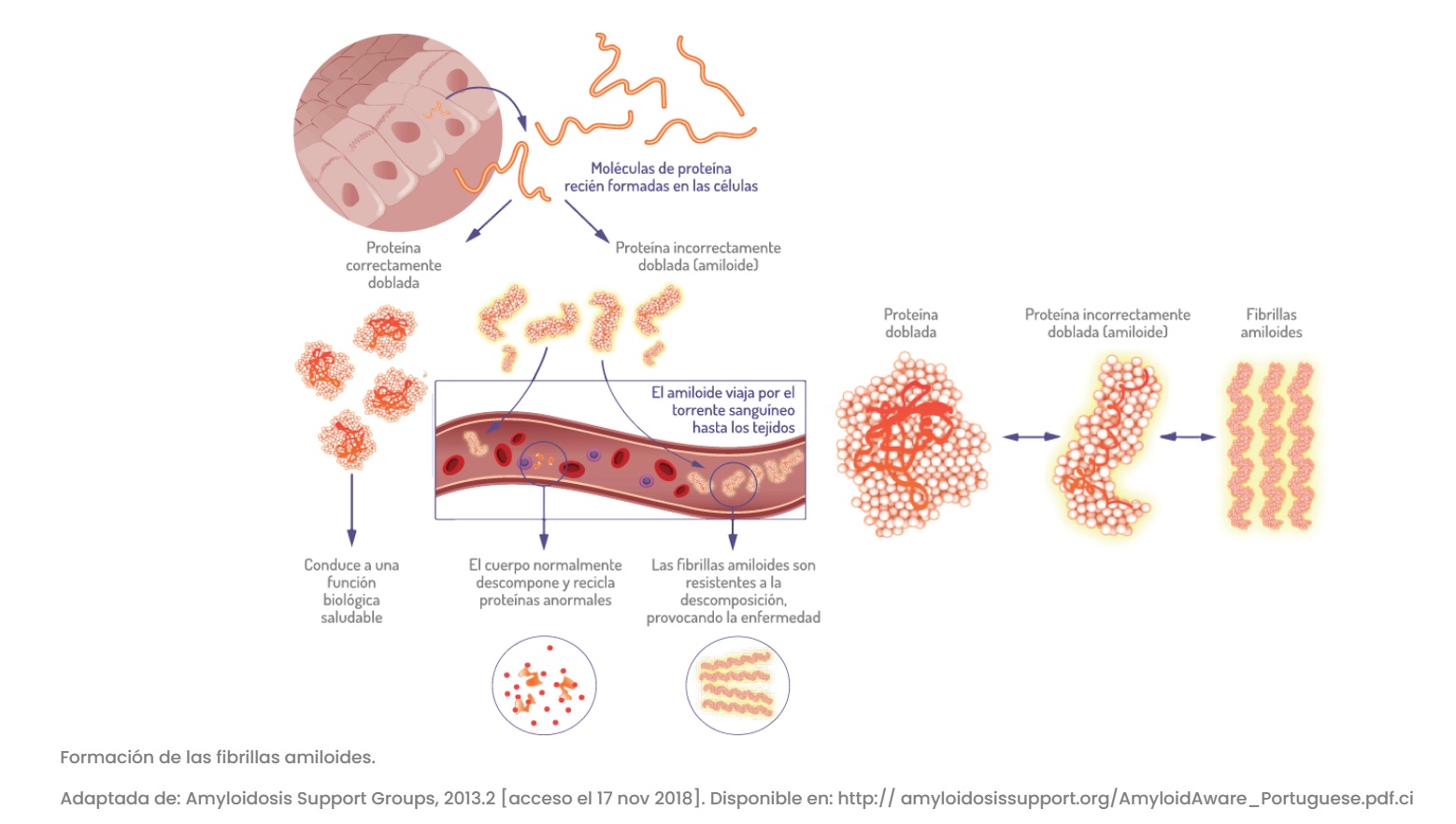
Una de las tantas EPOF que se busca concientizar este 2024 es la PAF-TTR (Polineuropatía Amiloidótica Familiar por TTR), también conocida como hATTR (Amiloidosis Hereditaria por Transtiretina) es progresiva y forma parte de un grupo de enfermedades llamadas “Amiloidosis”.

Basados en los datos epidemiológicos disponibles en Orphanet (1), se ha determinado que en el mundo hay descriptas clínicamente al menos 8.000 enfermedades poco frecuentes definidas por prevalencia puntual sin contemplar los cánceres raros, ni las enfermedades poco frecuentes causadas por enfermedades infecciosas o intoxicaciones bacterianas o virales raras. Por tanto, se reconoce que el número total es considerablemente mayor llegando a más de 10.000 EPOF. Cada año se agregan unas 300 nuevas descripciones
El mayor desafío es llegar a un diagnóstico certero, en ese sentido, en promedio, puede llevar entre 5 y 10 años, como también requerir hasta ocho visitas a distintos especialistas antes de obtenerlo. Se estima que más de cuatro de cada diez personas reciben al menos un diagnóstico errado durante el proceso.
Son enfermedades crónicas, complejas, progresivas, discapacitantes y, en ciertos casos, potencialmente mortales. Siete de cada 10 (72%) son de origen genético, y de ellas, el 70% se manifiesta al nacer o durante la niñez, siendo que 3 de cada 10 niños morirán antes de cumplir los 5 años. Estas enfermedades afectan entre el 4 y el 8% de la población mundial4, 5. E incluso, publicaciones recientes refieren que constituyen hasta el 10% de la población mundial: más de 300 millones de personas en el mundo viven con alguna enfermedad poco frecuente.
Qué es la Amiloidosis Hereditaria por Transtiretina
Este año se busca concientizar en Amiloidosis Hereditaria por Transtiretina, también conocida como Polineuropatía Amiloidótica Familiar, una enfermedad poco frecuente, hereditaria y progresiva, que afecta el sistema nervioso y distintos órganos del cuerpo. El nombre polineuropatía sugiere una enfermedad que afecta muchos nervios. Se llama amiloidótica porque su daño está asociado al depósito de fibras proteicas denominadas amiloides, que tiene impacto en la estructura y en la función de los tejidos afectados.
El principal obstáculo para diagnosticar esta patología es que sus síntomas son inespecíficos. Algunos de los síntomas más frecuentes incluyen mareos, diarrea, constipación, dificultades para caminar, hormigueos en las piernas. La afección clínica aparece alrededor de los 30 años o incluso después de los 60 años en algunos fenotipos. Sin embargo, la proteína defectuosa va afectando el cuerpo por goteo. Por eso, estos primeros síntomas pueden aparecer hasta 20 años antes de una manifestación más clara de la hATTR. Sin embargo el depósito de amiloide se va generando lentamente . Por eso los primeros síntomas no aparecen hasta la adultez.
Su sintomatología heterogénea y la poca difusión de información acerca de esta enfermedad genera que los pacientes pasen hasta por cinco especialistas para ser diagnosticados correctamente. Eso resulta en mucho tiempo sin los cuidados correspondientes y por lo tanto en un mayor desarrollo del padecimiento, profundizando los síntomas y deteriorando la calidad de vida de la persona.
Las señales de alarma deben prenderse ante la aparición de un conjunto de síntomas. El primero es la neuropatía sensitivo-motora progresiva. Si a esta condición se le suman 1 o más de los siguientes síntomas es recomendable consultar con un profesional.
Para César Crespi (MP115.409 / MN 124.765), médico hepatólogo del Centro de Referencia en Enfermedades Raras y de Dificultoso Diagnóstico (CERyD) del Hospital San Juan de Dios de La Plata, “si existe la sospecha, la confirmación patológica de depósito amiloide y el diagnóstico genético son altamente recomendados. Además, investigaciones adicionales como evaluación neurológica, cardíaca, autonómica y oftalmológica, pueden dar más seguridad para obtener el diagnóstico correcto”.
“Lo primero que se hace cuando llega un paciente es un buen examen físico y si tiene síntomas en los miembros inferiores, un electromiograma es uno de los estudios más importantes, el otro estudio de relevancia es el ecocardiograma (una ecografía del corazón), y también se puede solicitar un tilt test (poner al paciente en una camilla y cambiar de posición y ver cómo reacciona su sistema cardiovascular) si las manifestaciones clínicas lo ameritan”. César Crespi, médico.
“El test genético es un componente crucial para determinar la enfermedad. En pacientes con historia familiar o con un conjunto de síntomas que representen signos de alerta. En familias con una mutación conocida se pueden realizar test genéticos directos para esa mutación. Cuando se desconoce la mutación o cuando no hay historia familiar se debe realizar secuenciación completa del gen TTR para la identificación de la variante genética correspondiente”, puntualiza.







